Alpha and Beta Thalassemia
What is Thalassemia? Thalassemia is an inherited blood disease, causing one’s body to produce lesser red blood cells than normal. The red blood cells protein, also called “hemoglobin,” reduces in quantity and availability when thalassemia begins to manifest. Haemoglobins function as the body’s-manufactured proteins carrying oxygen to and fro to the human body system. Though […] Read More
Top Doctors For Alpha and Beta Thalassemia Treatments






Top Hospitals For Alpha and Beta Thalassemia Treatments



































Alpha and Beta Thalassemia
What is Thalassemia?
Thalassemia is an inherited blood disease, causing one’s body to produce lesser red blood cells than normal. The red blood cells protein, also called “hemoglobin,” reduces in quantity and availability when thalassemia begins to manifest. Haemoglobins function as the body’s-manufactured proteins carrying oxygen to and fro to the human body system. Though an uncommon disease, thalassemia mostly results in serious complications which may lead to death when left untreated. Though a person with thalassemia is likely to have a normal life expectancy, severe heart complications may arise before 30.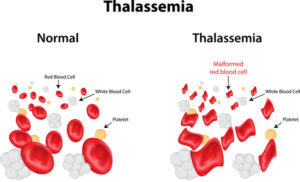 Picture Courtesy: medline
Picture Courtesy: medline
What are the most common types Thalassemia?
There are two main types of thalassemia, which include: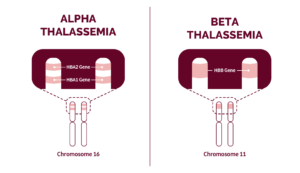 Picture Courtesy: Rare diseases
Picture Courtesy: Rare diseases
Alpha Thalassemia
This occurs when genes of alpha hemoglobin are mutated or reduced significantly. It involves the genes HBA1 and HBA2. In severe cases, this thalassemia is likely to affect the fetus in the mother’s womb, leading to the death of unborn babies due to the lack of oxygen supply and subsequent heart failure in the affected fetus. Alpha thalassemia is often caused by the abnormal or lack or loss of one to four genes. There are four types of this condition, and they include:-
-
- Silent Carrier; one gene missing.
- Carrier; two genes missing, usually occur with mild anemia.
- Hemoglobin (H disease), three genes missing, usually comes with a severe lack of blood.
- Alpha Thalassemia Major; all genes are missing, resulting in stillbirth or fetal death.
-
-
-
- Fatigue (tiredness and shortened breath)
- Jaundice (paleness of the skin and eyes)
- Swollen abdomen
- Darkened urine
- Abnormal slow growth
- Irritability
- Facial deformities
-
Beta Thalassemia
This happens when low levels of red blood cells lead to a lack of oxygen in many body parts. It is caused by the reduction or absence of the synthesis of beta-globin chains. Beta-globin is made up of two genes, one from each parent. Beta thalassemia is generally less risky and is caused by the absence of either one or both of the genes of one parent. The types of beta-thalassemia include:-
-
- Beta -Thalassemia Minor; is also known as the beta-thalassemia trait in which persons affected usually have a mild loss or lack of blood. It does not necessarily need treatment until serious symptoms occur.
- Beta-Thalassemia Intermedia; persons with this type of beta-thalassemia are prone to moderate lack of blood and would undergo blood transfusions.
- Beta- Thalassemia Major occurs when the parents’ beta-globin genes are missing. It is mostly recognized in the second year after the birth of an affected child.
-
-
-
- Severe lack of blood
- Paleness of eyes or skin–Jaundice
- Frequent infections
- Enlargement of body organs
- Moodiness
- Abnormal heartbeat
- Deformed bones.
-
What are the risk factors associated with Thalassemia?
A person falls at a higher risk of having any form of thalassemia if they:-
-
- Has a family history of thalassemia.
- Originates from Africa, Asia, or the Mediterranean.
- Has both parents with the disorder.
- Falls within the age range of 19-28 as an adult.
- Falls within the range of fetus to 2 years as an infant.
- Has sickle cell anemia.
-
What are the complications associated with thalassemia?
When thalassemia is left untreated, a lot of complications may arise. These include:-
-
- The accumulation of iron in the blood.
- Severe loss of blood.
- Abnormal changes in the bone structure of one’s body.
- Hampered growth, which would consequently lead to delayed puberty.
- Facial distortion is due to the abnormal shifts in the bone structure of one’s face.
- An enlarged spleen may lead to removing the spleen, thereby making the body prone to a higher rate of infections.
- Abnormal rhythms of the heart.
- Heart failure or other complications of the heart.
- Liver problems.
-
Diagnosis
As soon as the patient notices any symptom of thalassemia, they should seek the doctor immediately. The doctor will examine the patient and ask questions, which the patient has to answer correctly (to ensure safe results). Once the doctor suspects that the patient is suffering from the disorder, the patient has to undergo the following tests: Blood Tests. This would involve the examination of the blood. The doctor will perform any or all of the following tests, depending on the physical examination and history of complaints from the patient.-
-
- A complete blood count (CBC)test.
-
-
-
- A reticulocyte count.
-
-
-
- An iron test.
-
-
-
- Prenatal Tests.
-
-
-
- DNA testing/Genetic tests.
-
-
-
- Amniocentesis tests.
-
-
-
- Chorionic villus sampling.
-
What are the Treatment options available in treating thalassemia?
There are various ways of treating all forms of thalassemia. Some of these methods include the below:-
-
- The Use of Vitamins.
-
-
-
- Iron Chelation Therapy.
-
-
-
- Deferoxamine (injected into the skin).
- Deferasirox (oral medications ).
- Folate (folic acid) supplements.
-
-
-
- Blood Transfusion.
-
-
-
- Stem Cells /Bone Marrow Transplant.
-
-
-
- Growth Hormone Therapy.
-
-
-
- Genetic Therapy.
-
-
-
- Surgery.
-
Lifestyle Management
For persons living with thalassemia, proper healthcare is needed. As a thalassemic individual, the patient is advised to follow the instructions strictly. A few of the instructions are listed below:-
-
- Attend all appointments made by the physician.
- Honestly answer all questions asked by the doctor before undergoing any treatment procedure.
- Keep an attitude towards positivity; avoid negative thoughts about one’s health condition.
- Maintain a healthy and adequately balanced diet by:
- Avoiding cereals, bread, and juices.
- Eating plant-based foods.
- Avoiding iron-rich foods (if your iron level is high).
- Eating leafy foods and legumes (as they contain folic acids that fight off iron levels).
- Exercise regularly. Do not stress too much.
- Be in close contact with friends and support group networks.
- Seek prenatal counseling (if pregnant).
- Seek genetic counseling.
- Consult the doctor promptly whenever the patient has a fever or other illness.
- Avoid iron pills (since they contribute to the pileup of irons in the bloodstream).
- Wash hands regularly.
- Consult the doctor about supplements like: Calcium, Vitamins, Folates.
- Observe a safe distance from sick persons.
-
What is the life expectancy of a thalassemia patient?
Thalassemia is not a curable condition. It is a genetic disorder. A patient undergoing stem cells and bone marrow transplants can help treat the disorder. Children under the age of 10 have an estimated 99% survival rate from the condition. Upon clocking 20, young adults can live up to 30, with a survival rate of approximately 88%. After turning 30, thalassemic adults live at an estimated survival rate of 74% until they are 45 years old. Approximately 68% of thalassemic individuals can survive till the age of 50. Only an estimate of 51% of persons suffering from the disorder (thalassemia) can live up to 55. Women generally have a longer survival rate, while men have a higher mortality rate of 45%. As of 2015, approximately 17,000 people have died from thalassemia, bringing the number of deaths in 2021 to probably 29,000 individuals or more. The disorder has an approximate death rate of 7%.Symptoms
SYMPTOMS
When you happen to have thalassemia, you may observe the following symptoms:
- Slow or delayed growth in infants.
- Weak or deformed bones.
- Fatigue, drowsiness, weakness, tiredness.
- The enlargement of your spleen; Your spleen is an organ in your body that is responsible for purifying your blood and fighting off infections, in your abdomen.
- Pale or yellow skin or eyes (jaundice).
- Malnutrition or poor feeding.
- Problems about the loss of appetite.
- Irregular heartbeat.
- Chest pains.
- Heart failure or other complications of the heart.
- The dark shade of urine.
- Shortened breath or other breath-related issues.
- Headaches.
- Cold hands and feet.
- Leg cramps.
- High possibility of being prone to infections.
- Facial bone deformities.
- Excess iron in the bloodstream.
- Growth failure or stunted growth.
- Swelling of the stomach area.
Causes
CAUSES OF THALASSEMIA
Thalassemia is a genetic distorted trait passed down from parents to offspring. However, the cause of the condition depends on the type. Alpha thalassemia is caused by the absence of one to four of the hemoglobin genes, while beta-thalassemia is caused by the absence of the two beta-globin genes gotten from each parent.
FAQ
-
- Can thalassemia be cured?
Thalassemia is a genetic disease. There is no cure for it, but it can be suppressed with some specific treatments. Stem cells and bone marrow transplants can cure thalassemia, while other treatments like surgery and iron chelation can only reduce it.
A few individuals suffering from thalassemia major can get a good donor match and get their blood transfused, while many others do not.
Thalassemic patients who cannot get compatible blood from their donors may end up getting alloimmunized (a condition whereby a person’s blood may react to new blood from a donor and tries destroying it).
-
- How do I know if I have a thalassemia carrier?
The doctor will examine the patient suspected of having a thalassemia carrier by carrying out a few blood tests. The blood tests can take the form of a complete blood count, iron test, or reticulocyte count. The hematologist can perform these tests with the help of a functioning microscope.
-
- Is thalassemia a serious disease?
Thalassemia is a critical condition. Its signs and symptoms show up two years after birth. These pointers include pale skin, frequent loss of appetite, and stunted growth.
Routine blood transfusions and growth hormone therapies can serve as proper methods of treating thalassemia, but the frequency of blood transfusions can result in bad effects. Therefore, continuous blood transfusions are not medically advisable.
-
- Is thalassemia linked to leukemia?
According to the NHI (National Health Institute), the coexistence of thalassemia with cancers like leukemia has been reported. This coexistence could be explained by either habitational (relating to habitat or natural environment) or genetic relationships.
-
- Does thalassemia weaken the immune system?
Some of their bodies’ defenses against infections do not work when people suffer from thalassemia. When such happens, extra protection is needed.
Some of the preventive measures taken against thalassemia, which weakens the immune system and makes its patients prone to diverse infections, include:
-
-
- Covid-19 vaccines.
- Anti-malaria drugs.
- Flu shots and other vaccines.
- What is the difference between alpha and beta-thalassemia?
-
Thalassemia is a group of genetic disorders characterized by defects in the production of hemoglobulin. Thalassemia can be divided into two groups. They are Alpha thalassemia and Beta-thalassemia. Alpha thalassemia is caused by reduced or absent synthesis of alpha-globin, while beta-thalassemia is caused by reduced or absent synthesis of beta-globin chains.
