Multiple Endocrine Neoplasia
What Is Multiple Endocrine Neoplasia Type 1 (MEN 1)? MEN 1 is a rare genetic disorder characterized by multiple tumors emerging from cells of specific neuroendocrine tissues. Neuroendocrine cells are like neurons (nerve cells) but also make hormones like endocrine cells. They make and release hormones in response to signals they receive from the nervous system. These hormones control many body functions. Read More
Top Doctors For Multiple Endocrine Neoplasia Treatments




















Top Hospitals For Multiple Endocrine Neoplasia Treatments




































Multiple Endocrine Neoplasia
Table of contents
What Is Multiple Endocrine Neoplasia Type 1 (MEN 1)?
MEN 1 is a rare genetic disorder characterized by multiple tumors emerging from cells of specific neuroendocrine tissues. Neuroendocrine cells are like neurons (nerve cells) but also make hormones like endocrine cells. They make and release hormones in response to signals they receive from the nervous system. These hormones control many body functions. Hormones get involved in diverse vital and metabolic processes in the body, including:
- Regulating the heart rate.
- Blood pressure regulation.
- Refuting body temperature.
- Cell differentiation and growth.
In MEN type 1 patients, tumors develop in multiple endocrine glands, majorly the gastro pancreatic tract, parathyroid, and pituitary gland. This triggers the secretion of excessive amounts of hormones from the affected glands into the bloodstream, which results in varying symptoms and related syndromes. Some tumors, such as gastrinomas and carcinoids linked with MEN type 1, are malignant and can spread cancer to other body organs (metastases). Other manifestations of MEN 1, though not regular, are:
- Pancreatic neuroendocrine tumors
- Adrenocortical tumors
- Visceral leiomyomas
- Breast carcinoma
- Lipomas
- Facial angiofibroma
- Facial collagenomas
- Meningiomas
- Ependymomas
Multiple endocrine neoplasia type 1 can be inherited or occur due to a new gene mutation in the affected individual. MEN type 1 affects equally men and women. About 40% of MEN 1 cases involve tumors of the parathyroid, pancreas, and pituitary glands. Although it affects other ages, ages 20 to 40 are mainly affected. MEN 1 affects approximately 1 in 30,000 individuals.
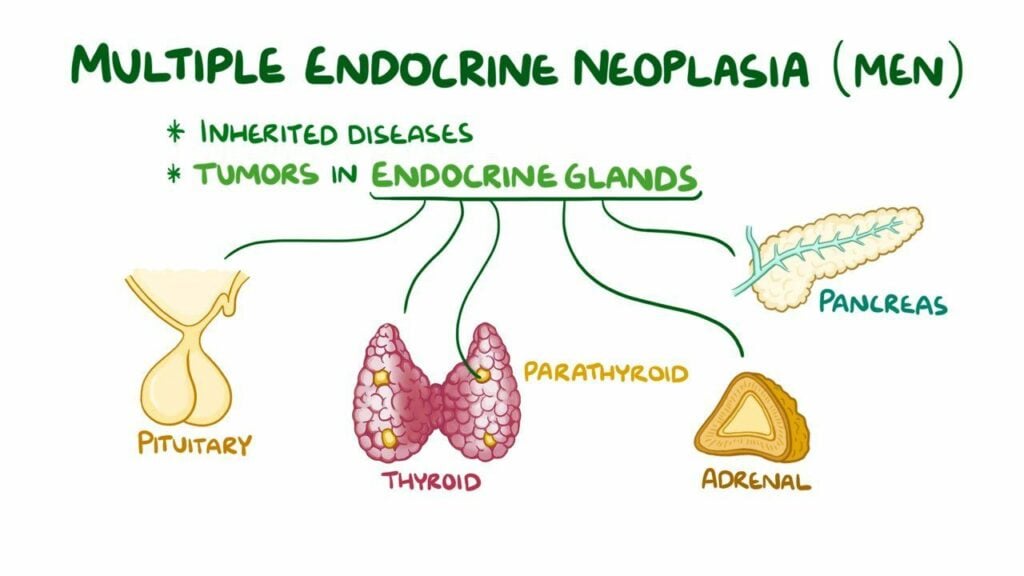
Causes
MEN type 1 is caused by the mutation in the gene of an affected person. MEN 1 is a tumor successor gene. It encodes a protein, menin, that is responsible for keeping cells from growing and dividing too rapidly or uncontrollably. >500 mutations of this gene have been identified.
Symptoms
The signs and symptoms of multiple endocrine neoplasia type 1 are several. It spreads all through the body and affects many parts and organs of the body quickly.
The signs should be detected and diagnosed at the earliest stage because once it begins to spread rapidly, the symptoms become complicated, and treatment is more difficult.
The signs and symptoms may include:
- Fatigue, tiredness, and depression.
- Headaches
- Body pains
- Pains in the bone.
- Visual disturbance.
- Diarrhea
- Peptic ulcers
- Pituitary tumors and other tumor formations in the body.
- Kidney stones
- Hyperplasia
- Acromegaly
- Cushing syndrome
- Galactorrhea
- Hypercalcemia
- Osteoporosis (broken bones).
How is MEN1 diagnosed?
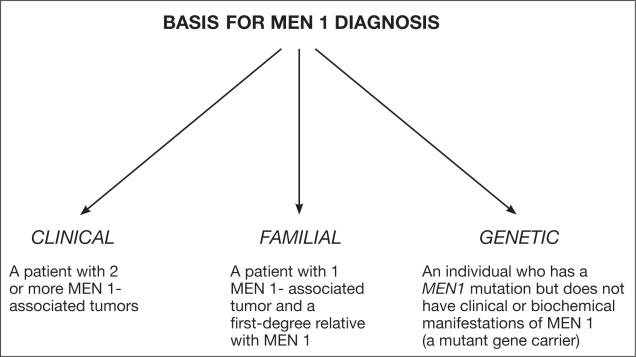
Diagnosis of MEN type 1 follows a thorough clinical evaluation.
- Doctor’s examination and findings
- Health experiences and history
- Family history
- Physical exams.
Details of diagnosis include identification of at least 2 or 3 characteristic endocrines (pancreatic, parathyroid, pituitary) tumors associated with the disorder and identification of an inactivating MEN 1 gene mutation.
- Lab tests such as blood and urine tests to determine hormonal levels in the body.
- Imaging tests to determine the location and size of specific tumors in the body. They include:
- MRI
- CT scan
- PET scan
- Nuclear medicine scan
- Endocrine ultrasound of the pancreas.
- Genetic testing to confirm the MEN 1 gene behind the disorder.
How is MEN1 treated?
The treatment of MEN 1 may require a team of combined efforts of medical specialists such as cancer specialists (oncologists), endocrinologists, surgeons, psychiatric specialists, and other healthcare professionals. Treatment may be directed toward the specific symptoms that are observed. Treatment options include:
- Surgical removal of the tumor
- Medical therapies such as SSAs, cytotoxic chemotherapy (streptomycin and 5-fluorouracil, doxorubicin, temozolomide with capecitabine)
- Peptide receptor radionuclide therapy.
- Inhibitors of thyroid kinase receptors (sunitinib).
- Inhibitors of mammalian target of rapamycin (mTOR or everolimus)
- Radiation therapy.
- Stereotaxic radiosurgery.
(Note: the medical team will decide which of the medical procedures to apply in careful consultation with the patient) The main treatment for parathyroid tumors is a surgery called a parathyroidectomy. This may be followed by the transplantation of healthy parathyroid tissue positioned in another area of the body. After a successful operation, the patient would be placed on a daily supplementation of calcium and vitamin D to prevent the effects of possible hypothyroidism. Neuroendocrine tumors may involve surgery (pancreatoduodenectomy or duodenectomy) or an ablation procedure. Metastatic neuroendocrine tumors may involve liver surgery, radiofrequency ablation, cryoablation, or chemoembolization.
What are the complications associated with MEN1?
Because of the rapidity and the widespread nature of the tumors in various vital organs and parts of the body, MEN 1 can cause a lot of life-threatening health risks. The complications include:
- Several cancerous tumors of different areas of the body.
- The nervous system breaks down.
- Kidney failure
- Heart failure
- Brain failure can lead to coma and brain death.
- Death.
FAQ
Multiple endocrine neoplasia type 1 is a genetic condition that produces tumors in the endocrine glands. Endocrine glands are the hormones producing the platform of the body. MEN 1 tumors mostly involve the parathyroid grads, islet cells of the pancreas, and pituitary glands.
Other alternative names for MEN 1 disease are Wermer syndrome, MEN 1 syndrome, and Multiple Endocrine Adenomatosis.
MEN 1 syndrome does not have a permanent cure because of its metastatic characteristics. Organs considered treated may redevelop tumors.
Surgery is often successful in removing MEN 1-related tumors and related symptoms. But these tumors, in some cases, may grow back or spread to the lymph nodes linked, the liver, or bones. The surgeon may give medications to reduce the size of the tumor as well as treatment-related problems.
MEN 1 is a rare genetic disease that affects both males and females equally. It affects about 1 in 30,000 individuals. However, there are other rarer types of multiple endocrine neoplasias; MEN 2, MEN 2A, MEN 2B, and FMTC. MEN 2affects about 1 in 35,000 people, while others are far lesser.
The high-risk factors and groups of MEN 1 include:
a) People who have gene mutations.
b) The persons within the range of 20 to 60 years.
c) People with health conditions such as:
d) Primary hyperparathyroidism (PHPT)
e) Zollinger-Ellison syndrome (ZES)
f) VIPomas
g) Kidney stones
h) Hypercalcemia
i) Osteoporosis
j) Gastrinomas
k) Prolactinomas
l) Glucagonomas and somatostratriomas.
If properly controlled, the median survival period for MEN 1 patients is 15 years. Men can live up to 55.4 years, and women for up to 47 years.
Pancreatic endocrine tumors (gastrinomas) become cancerous in half of the patients with MEN 1. Untreated patients may die from peptic ulcers, metastatic endocrine pancreatic carcinoma, or foregut carcinoid malignancy.
